
Frequently Asked Questions
- Why should I use this?
-
We realize that the there are many data processing options available, but also realize that not a single one
of them will perform ideally for each and every situation. By supplying the PeakML format and integration for
XCMS we try to achieve communication and exchange between different packages. For example, it is already possible
to do data extraction with XCMS and visualize the results in the PeakML Viewer and do all kinds of operations
on the data. With more supported software researchers will be able to cherry-pick parts from different packages
best suited to their needs.
Apart from this consideration, a well-defined and documented API is provided for creating custom-made tools. The choice of the Java programming language should enable a large portion of the bioinformatics community to plug into the development efforts.
- Why develop in Java?
-
The Java programming language offers the necessary language constructs (like generics and reflection) in order to
create a well-defined Application Programming Interface. The fact that the software is platform independent out of
the box helps with the quick implementation of tools for a wide range of platforms. Supported by libraries like SWT
and JFreeChart efficient and native looking user interface applications can be developed. This list of features is
encountered in other languages, but the fact that Java is a dominant language in bioinformatics additionally helped
make the choice. The memory consumption of Java admittedly is a big problem, which we have tried to circumvent by
storing data in efficient constructs and choosing to support centroid data alone at this point.
- Why yet another file format?
-
We tried to integrate the ideas into the mzML format, which has support for mass chromatogram data. However, this
format only supports single chromatogram elements (i.e. from a single measurement) and will not allow for the
integration of sets and extended meta information needed to accomplish the same functionality as the PeakML file format.
We decided that, instead of extending the already complex mzML format needlessly with more options and complexity, a
clean new approach was the best course to follow.
- Why use the SWT toolkit and not SWING?
-
The Standard Widget Toolkit provides platform specific implementations for user interface development. This means that
the user interface always looks and feels native for the platform on which it is run and is responsive. Unlike the SWING
library, SWT does have the consequence that special versions for each platform needs to be made by linking to the correct
SWT-library. But especially the native look and feel of the application makes this well worth the effort.
- PeakML Viewer: how do I zoom in/out on one of the graphs
-
Press the mouse on the upper-left corner of where to zoom and drag the mouse to the bottom-right corner and release the
mouse button. While dragging the mouse a rubberband will be drawn of the new area.
- PeakML Viewer: freezes when I try to zoom in one of the graphs
-
This issue only seems to affect 32-bit Windows XP installs so far. It appears that the standard javaw.exe (located in
the windows system directory) used in Windows is a different version then the highest installed java-version. In order
to rectify this, change the path to the javaw.exe of the highest installed version (likely to be found in
C:\Program Files\Java\) in the properties of the shortcut (right-mouse button on the shortcut file; click the item
properties).
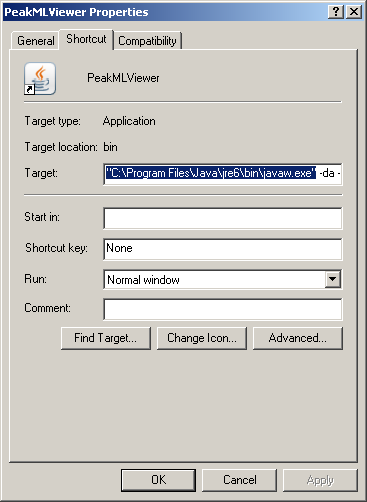
Be sure to put quotes around the path of the javaw.exe file.
- PeakML Viewer: freezes when I load a file
-
It is likely that the viewer was started by double-clicking on the peakmlviewer.jar file, instead of the shortcut
PeakMLViewer. The application always needs to be started with the shortcut, in order to have sufficient amounts of
memory available.
- mzMatch: ExtractMassChromatograms yields too much noise
-
The algorithm for locating mass chromatograms employs a na�ve approach, extracting every data point from a measurement.
This means that all of the real signals will be caught, but also a huge amounts of noise (e.g. in the case of the
LTQ-Orbitrap). The reason for this approach is to be sure that no reproducible signal is lost due to choices made for
their extraction. In order to remove a large part of the noise tools like mzmatch.ipeak.filter.RSDFilter and
mzmatch.ipeak.filter.NoiseFilter are provided. The rsd-filter is the most effective noise-removal mechanism, but it
does require a large amount of technical and/or biological replicates to be effective.
- mzMatch: freezes when I�m running my pipeline
-
It's likely that the pipeline is not started with the correct java options. The line below should be used for correct
operation:
java -da -dsa -Xmn1g -Xms1425m -Xmx1425m -Xss128k -XX:+UseParallelGC -XX:ParallelGCThreads=10 -cp mzmatch.jar
- PeakML: IXRawfile.open always results in RTCODE_RAW_FILE_INVALID
-
This error has been reported for a single machine, where the open function always resulted in this exception (even for
files which opened fine on different machines). A re-install of Xcalibur fixed this issue, but so far no satisfying
explanation has been found.